Imputing
imputing.RmdAlong with normalization, imputing missing values is another
important task in quantitative proteomics that can be challenging to
implement given the desired method. Again, tidyproteomics attempts to
facilitate this with the impute() function, which currently
can support any base level or user defined function along with
implementing the R package missForest, widely regarded as one
of the best algorithms for missing value imputation. Note that this
method is a matrix based sample imputation.
While random forest algorithms have shown superiority in imputation and regression, that does not portend their use in every case. For example, imputing missing values from a knock-out experiment, such as the dataset included in this package, are preferrable to minimum value imputation over the more complex random forest, simply because in this experiment we know how the experiment is affected.
Imputation in tidyproteomics attempts to apply each function
universally, meaning the same towards peptide and protein values. To do
this each data-object contains a variable called the
identifier , this tells the underlying helper functions
what values in the quantitative table “identify” the thing being
measured.
Proteins have a single identifier …
hela_proteins$identifier
#> [1] "protein"
hela_proteins$quantitative %>% head() %>% as.data.frame()
#> sample_id sample replicate protein abundance_raw
#> 1 9e6ed3ba control 1 Q15149 1011259992
#> 2 cc56fc1d control 2 Q15149 1093277593
#> 3 6a21f7a9 control 3 Q15149 980809516
#> 4 966be57f knockdown 1 Q15149 1410445367
#> 5 79a98e41 knockdown 2 Q15149 1072305561
#> 6 9f804505 knockdown 3 Q15149 1486561518Peptides have multiple identifiers …
hela_peptides$identifier
#> [1] "protein" "peptide" "modifications"
hela_peptides$quantitative %>% head() %>% as.data.frame()
#> sample_id sample replicate protein peptide
#> 1 74a86daf control 1 P06576 IPSAVGYQPTLATDMGTMQER
#> 2 74a86daf control 1 P06576 IPSAVGYQPTLATDMGTMQER
#> 3 74a86daf control 1 Q9P2E9 LTAEFEEAQTSACR
#> 4 74a86daf control 1 P11021 ITPSYVAFTPEGER
#> 5 74a86daf control 1 P11021 IINEPTAAAIAYGLDK
#> 6 74a86daf control 1 Q9P2E9 LLATEQEDAAVAK
#> modifications abundance_raw
#> 1 <NA> 43217337
#> 2 1xOxidation [M] 1465426
#> 3 1xCarbamidomethyl [C13] 5899490
#> 4 <NA> 52886668
#> 5 <NA> 244628414
#> 6 <NA> 5831700Imputation Functions
Imputation currently supports the following functions:
| Function | Method | Description |
|---|---|---|
| base::min | row or column | the minimum value in any given set |
| stats::median | row or column | the minimum value in any given set |
| user supplied function | any | e.g. function (x, na.rm) { quantile(x, 0.05, na.rm = na.rm)[1] } |
| impute.knn | matrix only | a non-linear KNN implementation (bioconductor::impute) |
| impute.randomforest | matrix only | a non-linear random forest implementation of missForest |
Imputing
rdata <- hela_proteinsAs part of this demonstration, signal from P06576 in p07_kd has been artificially removed to simulate a “genetic knockout mutation”.
w <- which(rdata$quantitative$protein == 'P06576' & rdata$quantitative$sample == 'knockdown')
rdata$quantitative <- rdata$quantitative[-w,]Using column
Note the difference using column ..
rdata %>%
impute(.function = base::min, method = 'column') %>%
subset(protein %like% "P23443|P51812|P06576") %>%
extract() %>%
ggplot(aes(replicate, abundance)) +
geom_point(aes(color=sample), size=3, alpha=.5) +
facet_wrap(~identifier) +
scale_y_log10(limits = c(1e4,1e9)) +
scale_color_manual(values = c('red','blue'))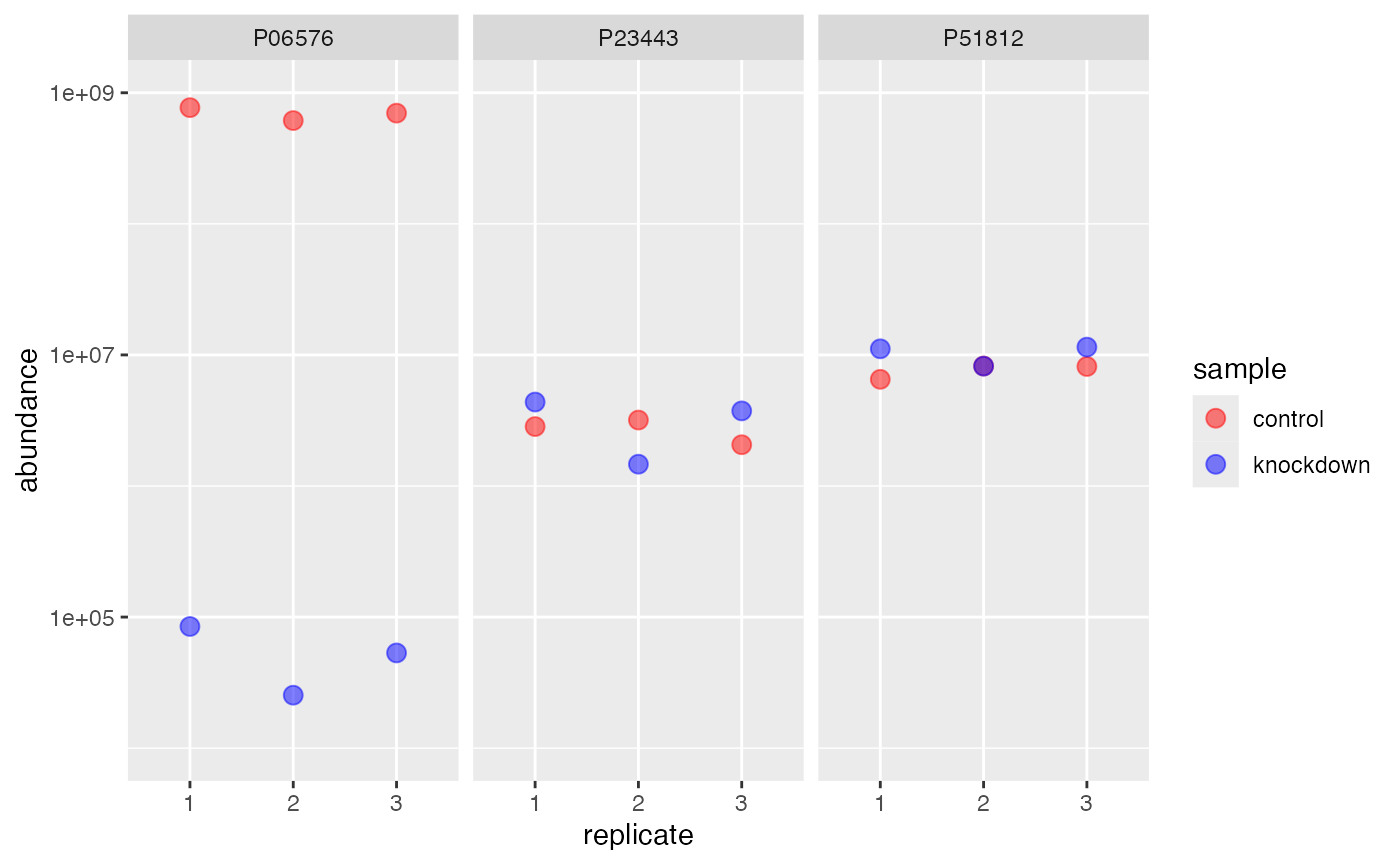
Using row
.. as opposed to row. The row method can be considered to contain the bias of any real offset, note our protein P06576 (i.e our artifical knock-out), shows the expected offset for the column method, and does not for the row method. Consider only using row methods when imputing values you suspect are missing-at-random. In our case P06576 is missing-not-at-random, because we performed a “genetic knockout mutation”.
rdata %>%
impute(.function = base::min, method = 'row') %>%
subset(protein %like% "P23443|P51812|P06576") %>%
extract() %>%
ggplot(aes(replicate, abundance)) +
geom_point(aes(color=sample), size=3, alpha=.5) +
facet_wrap(~identifier) +
scale_y_log10(limits = c(1e4,1e9)) +
scale_color_manual(values = c('red','blue'))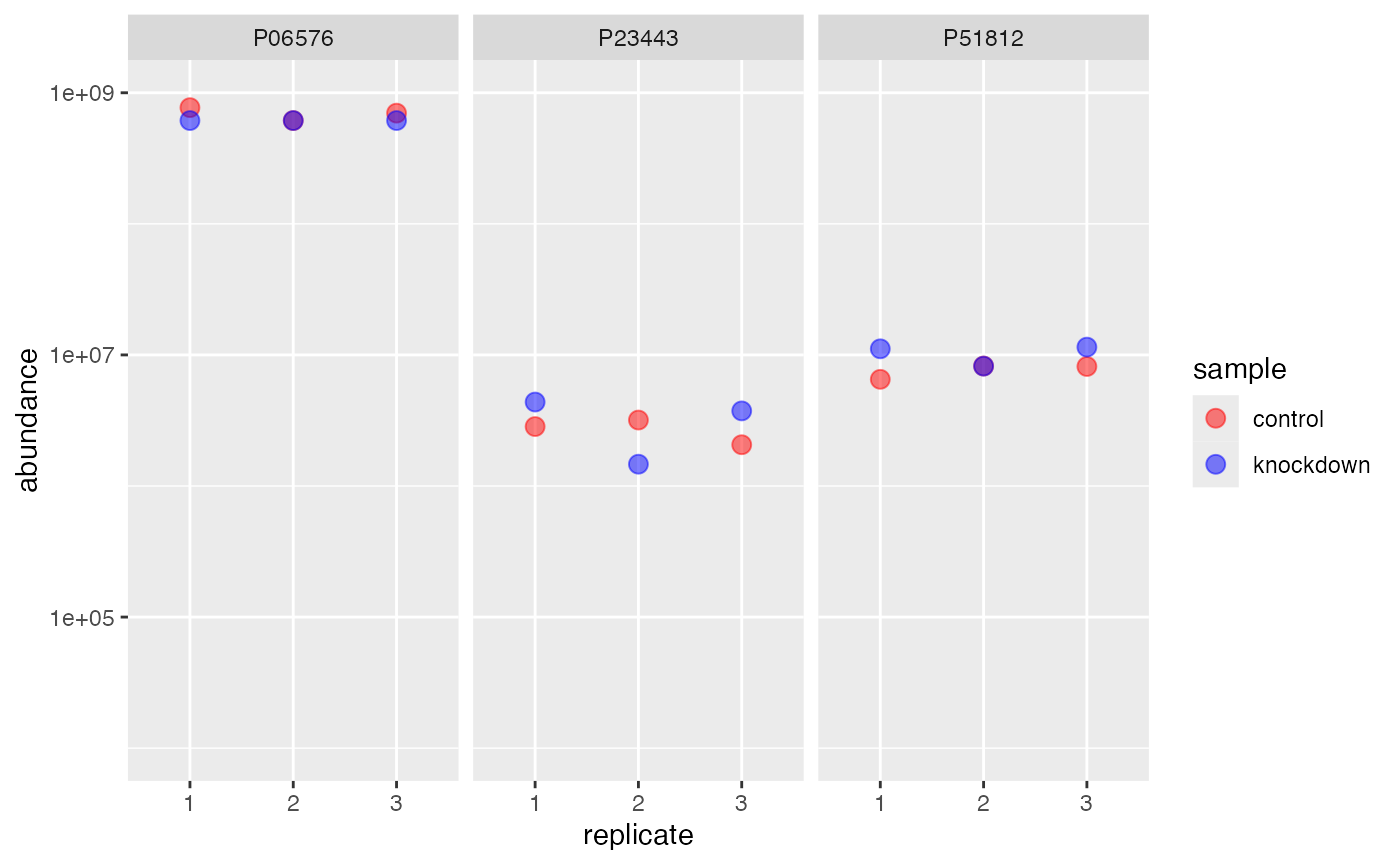
Using matrix
The matrix based operation takes advantage of data present in other samples (eg. “row”) and the information contained in the dynamic range (eg “column”) to better estimate the missing value - usually this is best for missing-at-random.
The R package in bioconductor::impute,
allows for the popular imputation method KNN. The generalized
impute function for the method matrix assumes
the underlying function is multithreaded, as is the impute.randomforest
method is. Therefore, to make any function operable, you need to make a
wrapper function to allow for the cores variable to be
accepted. In addition, the impute package returns an
incompatible data object, that you must convert to a matrix -
fortunately, the impute package’s data object contains the
matrix in $data.